
News from QED Bioscience
How to Obtain Stellar Staining with Fluorescent IHC
Related Antibodies from QED Bioscience
Catalog Name – Clone No.
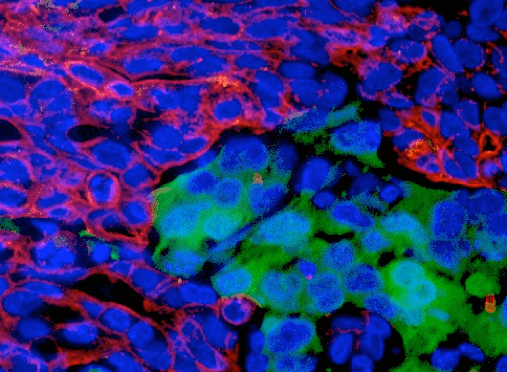
They say “a picture is worth a thousand words.” This is especially true in the case of fluorescent immunohistochemistry (IHC). With the flip of a microscope filter, you can gaze into a brightly colored galaxy of red, green, and blue pixels, waiting to be captured and presented in your next publication.
If you are interested in the kind of fluorescent IHC images worth those extra color publication charges, you’ve come to the right place. There are several important steps you can take to achieve dazzling fluorescent immunocytochemistry or IHC results.
1. Start with a Good Tissue Sample for Immunostaining
The two main methods for preserving tissues for IHC or immunohistology are paraffin embedding and freezing. While there is no universal preservation method suitable for detecting all antigens, the vast majority can be successfully detected in formalin-fixed paraffin-embedded (FFPE) tissue sections. For FFPE tissues, your original tissue sample should be thoroughly fixed, yet not over-fixed. Under-fixation leads to loss of tissue integrity, while over-fixation results in excessive crosslinking of your antigen of interest, making it inaccessible to your antibody during immunostaining. For small bits of fresh tissue (the size of a dime or less), overnight incubation in 4% paraformaldehyde, followed by transfer to 70% ethanol for long-term storage, works well. For other applications, perfusion with fixative is also satisfactory.
Some antigens, e.g. those with posttranslationally modified residues, will not survive even mild aldehyde fixation, and in such cases, the tissue should be snap-frozen and sectioned in a cryostat and stored at -80 °C until fixing in alcohol or cold acetone. Snap freezing prevents the formation of ice crystals. After fixation, these samples can be processed with standard IHC or immunohistology staining protocols. For frozen preparations, fresh tissues in optimal cutting temperature (OCT) compound or paraformaldehyde-fixed tissues in sucrose are snap-frozen in liquid nitrogen to prevent the formation of ice crystals.
Once your paraffin-embedded or frozen samples are ready, section them and affix them to slides for further analysis. And don’t forget, for a well-controlled study, you need both positive and negative tissue controls.
2. Deparaffinization and Rehydration
Once paraffin-embedded tissue sections are on slides, remove the paraffin by incubating slides in a series of xylenes and alcohols. Typically, this includes two passages in xylene, followed by three minutes each in 100%, 95% and 70% ethanol. To complete the rehydration process, wash sections two times in dH2O for 5 minutes each.
3. Antigen Retrieval
Deparaffinized formalin-fixed tissues generally require an antigen retrieval step. This helps remove crosslinks that may have developed during fixation so that your antibodies can more easily reach their target epitopes. There are citrate and Tris-based antigen retrieval buffers, while some methods call for treatment with enzymes such as pepsin, trypsin, or proteinase K.
Sodium citrate, pH 6.0, is the most commonly used antigen retrieval solution. Slides are brought to a boil in 10 mM sodium citrate buffer, pH 6.0, and maintained at just below boiling temperature for 10 minutes. Cool slides on bench top for 30 minutes. It is important not to over- or under-heat samples as this can cause inconsistent immunostaining results, so the heating process should be optimized with blank slides for your lab’s make and model of microwave as follows:
Heat blank slides until the buffer starts to boil. Adjust the power setting of the microwave to 30%, for example. Heat for 10 minutes and observe. A vigorous boil should be avoided. If a rapid boil is reached, remove the slides and repeat the optimization with fresh, room temperature buffer. Reduce the power to 20% instead of 30% and observe. Once the heating protocol has been optimized it should be recorded and always performed the same way. For example, if the heating protocol is optimized with 24 slides, then 24 slides should always be used (even if blank slides need to be used).
4. Permeabilization and Blocking Steps
Permeabilization is often required in order to stain nuclear or cytoplasmic antigens. Once your tissue sections have cooled, permeabilize them for about 5 minutes in a dilute detergent solution, such as Tween-20 or Triton X-100. This treatment will disrupt cell membranes in your tissue and allow antibodies to reach intracellular targets. Rinse slides with PBS.
After permeabilization, include a 10–15 minute blocking step with normal serum to prevent non-specific binding of primary and/or secondary antibodies. Antibodies and other proteins in blocking serum bind non-specifically to tissue sections and effectively block primary and secondary antibodies from binding non-specifically in later steps. Blocking serum can be the same species as that of the secondary antibody or of an unrelated species. However, it is very important to avoid any blocking solutions that contain serum that is of the same species as the primary antibody source because fluorescent secondary antibodies can bind and produce non-specific immunostaining.
5. Incubation with Primary Antibodies for IHC or Immunocytochemistry
Choose your primary antibody based on its specificity for your antigen of interest and its suitability for IHC or immunocytochemistry. Not all primary antibodies are created equal. Antibodies that are designed for western blot or ELISA assays may not recognize their target antigens that have been crosslinked in fixed tissue. Look for examples of fluorescent IHC or immunohistology results on commercial antibody data sheets as a good indication that the antibody will meet your needs. Even better, some data sheets have a recommended IHC and immunocytochemistry concentrations for primary antibody, a good starting point for your own experiments. Primary antibodies against multiple targets can be combined into one incubation solution, provided that they are of different host origins so that they can be detected with different secondary antibodies.
Once you have your primary antibody diluted (in PBS or PBS-Tween), incubate your sample for one hour at room temperature or overnight at 4ºC. For each experiment, include a tissue section incubated with a non-specific isotype control antibody that matches the class and type of the primary antibody (if the primary antibody is a monoclonal antibody), but does not recognize the target epitope. Isotype controls help distinguish non-specific background fluorescence from specific fluorescent labeling of your target antigen. If the primary antibody is a polyclonal antibody, include a tissue section incubated with a non-specific, species-matched polyclonal antibody.
6. Rinses
For most fluorescent IHC experiments, a simple PBS solution works well for rinses between antibody incubations. In some cases, 0.05% Tween-20 can be added to help reduce background, but it is usually not necessary.
7. Secondary Antibodies
Secondary antibodies can be conjugated to different fluorophores ranging from green to red to blue depending on the fluorescent conjugate. Choose your secondary antibody for its specificity to your primary antibody and the fluorescent color you desire. For example, a primary antibody raised in rabbit requires a secondary antibody against rabbit. Use multiple colors on one section by using secondary antibodies linked to contrasting fluorophores that distinguish between your primary antibodies based on host origin. Just as with primary antibodies, combine groups of secondary antibodies into one incubating solution. For the secondary antibodies, incubate for 45 minutes at room temperature in the dark to protect fluorophores from bleaching.
8. Nuclear Counterstain and Mounting Medium
A fluorescent nuclear counterstain is helpful for identifying cells in your sample that do not express your antigen of interest. A good counterstain may also provide some contextual and structural cues within your tissue sample. If you have not used the blue channel for one of your primary antibodies, incubate your sample in 1µg/mL DAPI (4′,6-Diamidino-2-Phenylindole), which labels cell nuclei, for five minutes. You can also purchase mounting medium that already contains DAPI to save yourself that extra step.
To mount coverslips to slides, use mounting medium designated for fluorescence microscopy to help preserve the fluorescent signal in your sample. Apply the coverslip with enough mounting medium, taking care to eliminate bubbles that can distort or obscure your images.
9. Visualization
Allow your slides to dry after coverslipping (if you can wait that long!) and visualize fluorescence using the filters on your microscope that are appropriate for each fluorophore. You can acquire images of the same microscopic field with multiple filters and merge the images later for a multi-color effect. Sometimes, the results are best seen in single fluorescent images placed side-by-side. If your tissue is thick, a confocal fluorescent microscope can be used to create z-stacks for clearer images that can be rotated for additional cues about antigen localization in three dimensions.
With careful planning and execution, you can achieve fluorescent IHC results that are both scientifically significant and visually stunning. Good luck!
View all IHC Antibodies >
Duraiyan J, Govindarajan R, Kaliyappan K, Palanisamy M. (2012) Applications of
immunohistochemistry. J Pharm Bioallied Sci. 4(Suppl 2):S307–9.